

 Mark Hammar
Mark Hammar Note: This article was updated according to the ISO 13485:2016 revision.
ISO 13485 is the international standard requirement for a medical device quality management system. Like many other quality management system requirements for special purposes (such as IATF 16949 for automotive production and service parts and AS9100 for use by aviation, space and defense organizations), the ISO 13485 standard is based on the requirements of ISO 9001.
Just like these other standards, ISO 13485 includes the entire ISO 9001 standard with additional requirements included in blue italics text. One major distinction of ISO 13485 is that it is intended to also be required for regulatory purposes as well as a non-statutory requirement for a quality management system.
What is ISO 13485 based on?
Until the introduction of the high-level structure in 2015, ISO 9001 and ISO 13485 were very similar standards and it was very easy to build a quality management system that met the requirements of both of them. However, today the situation is slightly different because ISO 13485:2016 still supports the structure of old ISO 9001 revision (from 2008).
The question everyone is asking is why 13485:2016 is so different from 9001:2015, when 13485:2016 came out just 6 months after 9001:2015 was released. Unfortunately, corrections on the ISO 13485:2003 took a long time. The new version of ISO 13485 was ready to be released in 2016 although it relied on ISO 9001:2008. So, when ISO 9001:2015 was finally released with the new structure, 13485:2016 was also already ready for release with the ISO 9001:2008 structure. The ISO organization decided to release 13485 with the old structure.
For information on the differences between ISO 9001:2015 and ISO 9001:2008, see Infographic: ISO 9001:2015 vs. 2008 revision – What has changed?
ISO 9001:2015 vs. ISO 13485:2016 – How are they similar?
Besides these differences in the structure, there are also similarities between ISO 9001:2015 and ISO 13485:2016:
- Risk based approach: Both standards emphasize the need to approach both production and business from a risk perspective and to make important decisions based on a risk analysis.
- Process approach: Both standards use the Plan-Do-Check-Act (PDCA) process approach.
- Customer Focus: Both standards are built around ensuring that customer requirements are met.
- Infrastructure: Both standards require identifying the infrastructure necessary for business processes.
- Employee Competency: Both standards require an organization to determine the competence of employees so that they can do the work assigned to them in accordance with regulatory requirements.
- Role of organization: Both standards require defining employee roles in the organizational structure.
In ISO 13485:2016, the manufacturer must show compliance with regulatory requirements in 58 places. For comparison, in ISO 9001:2015 the term ‘’regulatory requirements’’ is only mentioned 11 times.
What are the additional requirements in ISO 13485:2016?
After some information added in the introductory section that mainly tailors the text to the medical device industry, the first inclusions are in the terms and definitions. There are 14 new terms used by the medical device industry. The differences between manufacturers, importers, distributors, and terms such as clinical evaluation and post-market surveillance are specifically explained.
Listed below are additional requirements for ISO 13485:2016 when compared to ISO 9001:2015:
Clause 4 – Quality Management System
- Actions required to maintain effectiveness of the QMS (section 4.1)
- Changes to QMS documentation must be evaluated for their impact and controlled (section 4.1.4)
- Control and management of outsourced processes (section 4.1.5)
- Documented procedure for software validation (section 4.1.6)
- Documentation requirements per regulations (section 4.2.1)
- Quality manual, to include scope of the QMS and outline of documentation used (section 4.2.2)
- Documented file for each medical device maintained by the organization (section 4.2.3)
- Document controls to review and approve prior to use; re-approval by a designated function with pertinent information and control of obsolete document to include at least the lifetime of medical device it pertains to (or specified by law) (section 4.2.3)
- Records maintained for at least the lifetime of the medical device or as specified by law (section 4.2.4)
Clause 5 – Management Responsibility
- Management commitment to ensure effectiveness of the QMS (section 5.1)
- Management Rep to promote awareness of regulatory requirements (section 5.5.2)
- Management review to include review of revised regulatory requirements (section 5.6.2) and output to identify improvements for QMS effectiveness (section 5.6.3)
Clause 6 – Resource Management
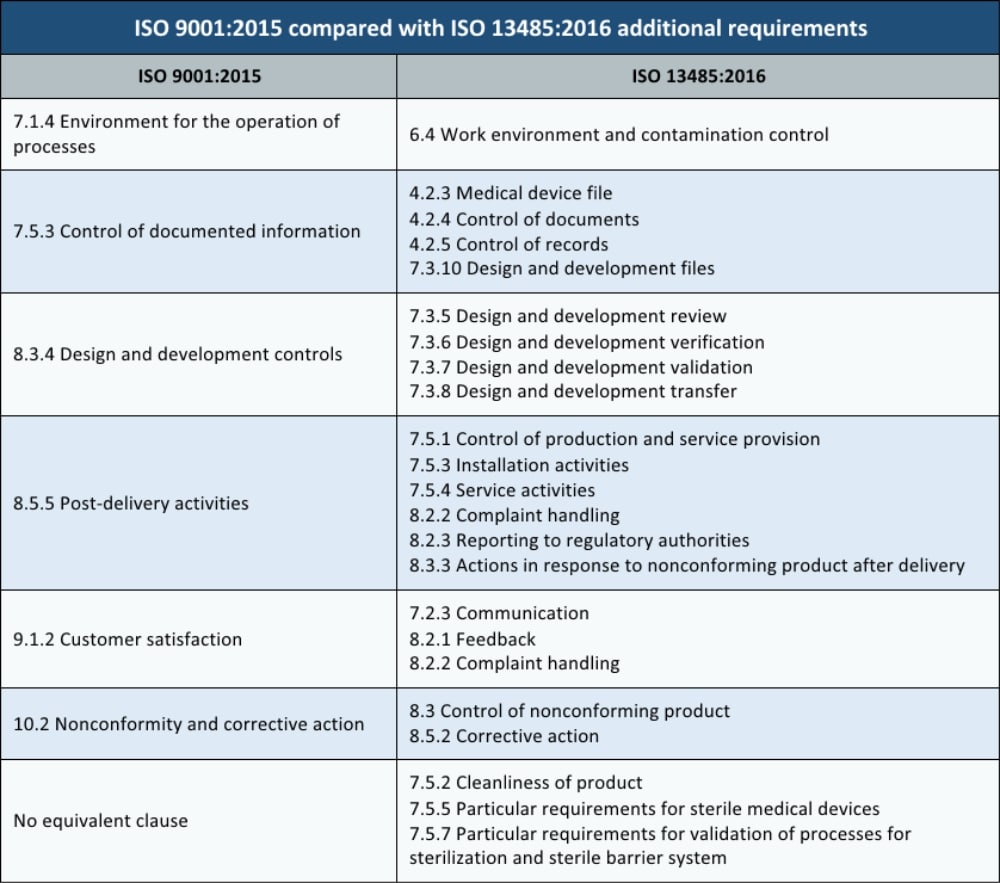
- Requirements for work environment, including cleanliness of clothing, temporary work conditions, and contaminated product controls (section 6.4)
Clause 7 – Product Realization
- Procedures for risk management associated with product realization planning (section 7.1)
- Advisory notices added to customer communication channels (section 7.2.3)
- Design and development procedures need to be documented (section 7.3.1). Additionally, throughout the subsections of section 7.3 on design and development, there are requirements relating to function, performance and safety requirements, risk management and the need for clinical evaluations.
- Throughout the subsections of section 7.5 (production and service provision), there are many specific medical device requirements that stress the need to follow regulations, including: cleanliness of product and contamination control, installation activities, servicing activities, requirements for sterile medical devices, documented procedures for validation of sterilization processes, identification and traceability requirements, status identification and additional product preservation information.
Clause 8 – Measurement, Analysis and Improvement
- Documented procedure required for a feedback system to establish if the company has met customer requirements (section 8.2.1)
- Additional requirements on monitoring and measurement of product include identification of the personnel performing inspections (section 8.2.4)
- Inclusion of the acceptance of a non-conformance only if regulatory requirements are still met, and control and authorization of rework instructions (section 8.3)
- Feedback included in analysis of data (section 8.4)
- The section on improvement general requirements includes the effectiveness of the QMS and record of customer complaints, including those not followed by corrective actions and notification when required by national or regional regulations (section 8.5.1)
- Including update of documents in the actions taken for corrective actions (section 8.5.2)
- Including records of investigations for corrective and preventive actions (section 8.5.2 & 8.5.3)
- Including effectiveness in the review of corrective and preventive actions (section 8.5.2 & 8.5.3)
If your organization is at all involved in the medical device industry, ISO 13485 is the QMS standard you should look at for additional requirements above and beyond ISO 9001.
To implement ISO 13485 easily and efficiently, use our ISO 13485 Documentation Toolkit that provides step-by-step guidance and all documents for full ISO 13485 compliance.




