

 Waqas Imam
Waqas Imam The requirements for medical device files in ISO 13485:2016 are an endeavor by the ISO Technical Committee (TC 210) to create consistent operations for medical device manufacturers, and also to make their Quality Management Systems compliant with the rules of various regulatory bodies.
Manufacturers and suppliers of medical devices must manage hundreds, if not thousands of different medical products. Therefore, it is vital that these organizations document their design and development processes, flow of manufacturing processes, records of devices, and advanced manufacturing controls for each device family. This allows the company to reproduce the processes again and again when needed, and to avoid any gaps or non-conformities during manufacture or delivery of a medical device. This is why ISO TC 210 includes the requirement of a medical device file.
Medical device files were not mandated in the previous revision of the standard, ISO 13485:2003, though it was a regulatory requirement in several countries – for example, FDA 21 CFR Section 820 in the United States, and Medical Device Directive 93/42/EEC in Europe. With the addition of Sub-clause 4.2.3 regarding Medical device files in ISO 13485:2016, the standard has brought increased value for implementing organizations.
Requirements of medical device files
Medical device files are documents that includes descriptions of design records, manufacturing processes, product specifications, device usage guides, quality measurement criteria, levels of compliance with regulatory bodies and quality standards, and, if required, servicing and installation records and their guidelines. Organizations should develop and maintain a medical device file for each product type or device family. Sub-clause 4.2.3 of ISO 13485:2016 sets requirements for various elements that should be incorporated in the medical device file. These elements include:
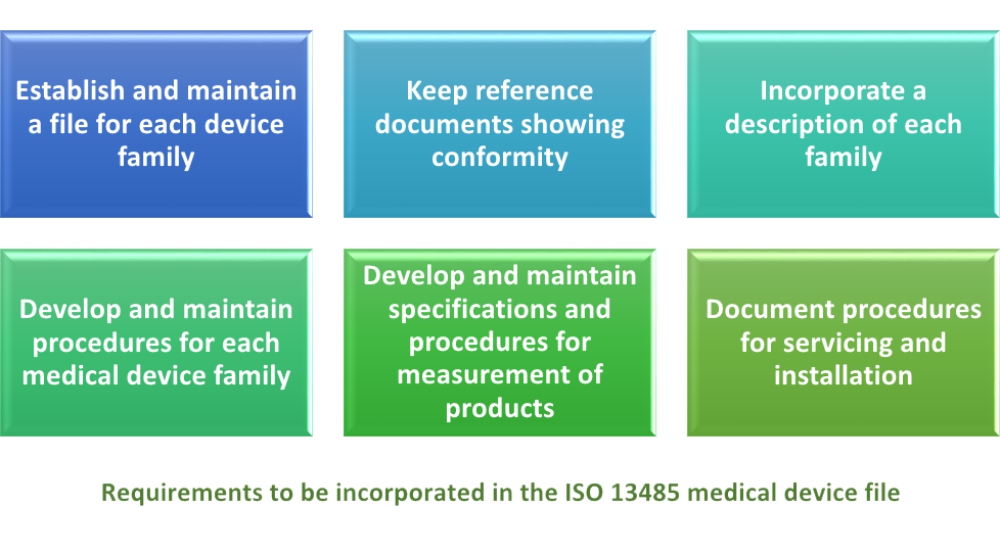
1) Establish and maintain a file for each device family – It is vital to understand a medical device family; for instance, we have Kelly forceps, which could be considered a medical device family. However, within this family there are various types of forceps that can differ in size, clamping specifications, handle specifications, material specifications, or other design specifications. But, the basics of design specifications can be grouped together as one family. So, for each group of devices that are considered to be a family, there must be a medical device file.
2) Keep reference documents showing conformity – For each medical device file, the organization should maintain references with their Certificate of Conformity to ISO 13485 and applicable regulatory requirements. This means that the medical device file should either contain the certificate of conformity, or it should refer to any document that proves that all processes in the development, manufacturing, packaging, storage, and handling conform to the requirements of ISO 13485 and applicable regulatory requirements. The reference can be a Quality Manual that is based on ISO 13485 and relevant regulatory requirements.
3) Incorporate a description of each family – The medical device file of each product family should incorporate a general description of the medical device, along with its intended use or purpose. It should also contain the master records for IFUs, i.e., instructions for use. The description should also include requirements for labeling, for example part code, device name, bar code, or CE requirement.
4) Develop and maintain procedures for each medical device family – Each medical device file must contain developed procedures, or specify procedures for production and all associated manufacturing processes, i.e., packing, inventory keeping, safe movement, and shipment of packed medical products. All manufacturing process flows, including the inspection points, for each medical device family need to be documented clearly.
5) Develop and maintain specifications and procedures for measurement of products – The medical device file should contain or refer to documentation of all specifications (e.g., device critical dimensions, material specifications, manufacturing specifications, and finishing specifications) for each SKU (Stock Keeping Unit). It should also specify the procedure for inspection of devices in this family, the checkpoints in the processes, the critical parameters of the products, and which instruments will be used to inspect critical parameters.
6) Document procedures for servicing and installation – This requirement is exclusive to devices or services that require servicing or installation, for example electrocardiographs, X-ray machines, etc. The medical device file should maintain, where applicable, documented information for installation of a device and should specify the procedure for servicing. Documented information for installation of a device may contain steps for installation, or installation records. The procedure for servicing can include the frequency of routine maintenance, and the process flow for preventive maintenance and repairs.
A foundation for manufacturing, delivery, and use of medical devices
The concept and requirements of medical device files serve as the foundation for the manufacturing process, delivery, and use of medical devices, as they detail all manufacturing and functional aspects of the medical product lifecycle. Before the requirement of the medical device file, organizations had to comply with different sets of documentation for each aspect of the product lifecycle, or they had to organize something similar to the medical device file; otherwise, they would have faced serious gaps in manufacturing process flow.
Medical device files minimize the production time, prevent duplicating processes, and minimize shipment damage within the manufacturing and delivery processes that can occur due to unclear or ambiguous manufacturing models. Organizations can effectively manage their manufacturing and shipment processes with the help of clear, documented procedures that are all centralized and controlled in a medical device file for each product device family.
To comply with all ISO 13485 requirements, use this helpful ISO 13485 Documentation Toolkit that provides QMS documents for medical device companies.



