

 Kristina Zvonar Brkic
Kristina Zvonar Brkic For each medical device that is to be sold in the European Union market, the manufacturer must prepare the technical documentation to demonstrate that the device was designed and manufactured in accordance with all applicable Medical Device Regulation (MDR) requirements.
What is post-market surveillance in the MDR?
Post-market surveillance is a process of proactively collecting and analyzing experience gained from devices on the market. This surveillance is crucial because some risks may appear only after use, in transport, during storage, or while cleaning.
However, the question is how to monitor what happens with the product after its use has started, whether there are situations or risks that were not foreseen, and whether any product improvement is needed. Given the large number of counterfeits that can be found on the EU market, the great competition for most medical devices, and the fact that the EU market is very large, during the preparation of the MDR, the EU commission realized the importance of a well-designed monitoring system once the medical device goes into circulation. That is why the post-market surveillance (PMS) system has strict and detailed requirements in the MDR.
What is post-market surveillance in MDR?
Post-market surveillance is a process of proactively collecting and analyzing experience gained from devices on the market. This surveillance is crucial because some risks may appear only after use, in transport, during storage, or while cleaning. Therefore, we can say that PMS serves to identify problems in the design and/or use of the device after it is placed on the market.
Requirements for PMS can be found in the following regulation and standards:
| Regulation/standard | Requirements for Post-Market Surveillance |
| Medical device regulation (MDR) 2017/745 |
|
| ISO/TR 20416:2020 Medical devices | Post-market surveillance for manufacturers |
| ISO 13485:2016 Medical devices | Quality management systems — Requirements for regulatory purposes, Clause 8 |
| ISO 14971:2019 Medical devices | Application of risk management to medical devices, clause 10 |
What is the purpose of post-market surveillance?
The purpose of a well-established PMS system is the timely identification of risks that were not known until use, as well as opportunities for product improvements, all with the aim to:
- ensure the safety and intended use of the medical device
- meet applicable regulatory compliance
- contribute to the lifecycle management
In the table below, you can see examples of the processes and activities providing inputs to post-market surveillance and the decisions or actions to take based on the post-market surveillance process.
Post-market surveillance processes and decisions
| Processes/activities providing inputs to PMS | Decisions or actions to take based on the post-market surveillance |
| Design and development | Design and development changes (design changes required by the user / patient – e.g., increase the diameter of the digital thermometer belt for obese people) |
| Risk management | Maintenance of the risk management file (any customer complaint or market information from the competition should be checked to see if it is covered by the manufacturer’s risk analysis, whether the specific risk is analyzed and controlled) |
| Clinical and performance evaluation | Maintenance of the clinical evaluation file (any new information related to the safety of the product and its purpose should be analyzed in the Clinical Evaluation Report) |
| Regulatory compliance | Reporting of adverse events (in case there is a serious incident you have to report to the regular authority) Issuance of an advisory notice (advisory notices are issued to distributors, users, and sometimes the general public through regulatory bodies – read more here: How to manage recalls and advisory notices for medical devices according to ISO 13485) |
| Improvement | Corrective and preventive actions (sometimes analyzing risks and complaints, and other information from the market, can lead to the need for changes within the quality system itself and, thus, by initiating corrective and / or preventive actions) |
| Marketing and sales | Maintenance of the marketing strategy |
This table shows that information gathered from post-market surveillance can be implemented in several elements of the Quality Management System. Furthermore, if the collected data from PMS changes the risk analysis and clinical evaluation, then it means that PMS has a direct impact on the change of technical documentation for the medical device.
For more information, read: What are the EU MDR technical documentation structure and requirements?
Who is responsible for post-market surveillance?
According to Article 15, the person responsible for regulatory compliance (PRRC) is responsible for post-market surveillance obligations. The manufacturer must nominate this person according to the competences stated in this same Article 15.
In ISO/TR 20416:2020, chapter 5.4 Responsibilities and authorities states that top management must define the post-market surveillance team. This team should include cross-functional representatives – process owners, for example, from the design and development department, risk management, quality assurance department, complaint handling department, and so on. The number of people involved in the PMS depends, of course, on both the size of the company and the complexity and risk of the medical device. Each process owner is responsible for collecting and analyzing data from his/her department.
Furthermore, in Annex C of ISO/TR 20416: 2020, point C 2.4 Responsibilities and authorities, the following term is introduced: post-market surveillance process owner, who is responsible for coordinating the post-market surveillance activities. This person is basically the PRRC when compared to the definition stated in Article 83 – the duty of the PRRC is to ensure that the PMS system is implemented and kept up to date.
To conclude, top management must nominate the PRRC and the post-market surveillance team. The PRRC then coordinates process owners for different post-market surveillance activities.
How do you conduct post-market surveillance?
According to clause 8 of ISO 13485:2016, and clause 10 of ISO 14971:2019, the manufacturer must document one or more processes for collecting and analyzing data from production and post-production activities. MDR Article 10, point 9. i) states that the manufacturer’s Quality Management System must address setting up, implementation, and maintenance of a post-market surveillance system in accordance with Article 83.
Therefore, first, it is necessary to prepare the documented procedure for PMS in which you will describe how your PMS system is organized. A well-established PMS system must define the following:
- which data from the market and public are necessary to follow
- methods for collecting those data
- methods for analyzing those data
- how to draw conclusions based on the analyzed data
- how to implement those conclusions into both quality management documentation and technical documentation (e.g., risk analysis, clinical evaluation, trend data analysis…)
The next thing is that top management must document the nomination of both the PRRC and the post-market surveillance team. The PRRC must prepare the PMS plan and organize a post-market surveillance review meeting with the post-market surveillance team. During this meeting, results and outcomes of different post-market surveillance activities are reviewed and discussed, and an overall conclusion is drawn.
Post-market surveillance plan. The PMS plan defines how the manufacturer intends to actively collect and analyze relevant data. The PMS plan must have the following data:
- Scope for the plan
- Objectives
- Responsibilities and authorities
- Data collection
- Data analysis
- Report on data analysis
- Review of the PMS plan
The PMS plan must be prepared before placing the medical device on the market for the first time, and updated as necessary during its lifecycle. This means that, if you have a new device, for first-time certification you need to have prepared the PMS plan, which is then a part of your technical documentation.
Post-market surveillance reporting
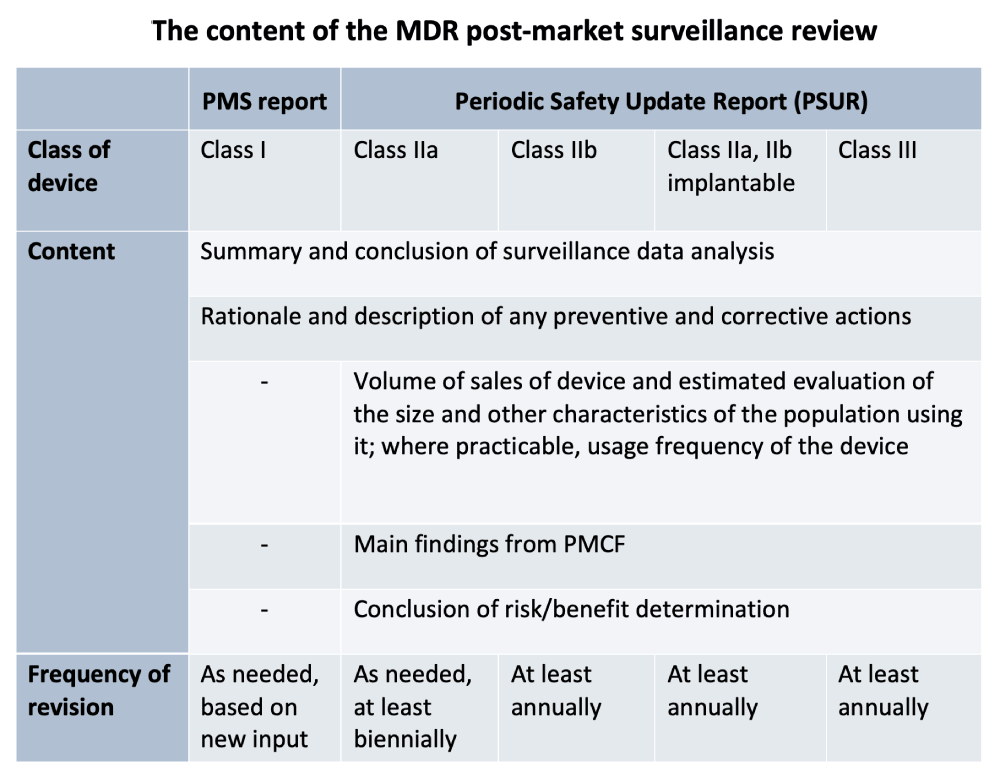
The MDR distinguishes between two types of reporting, depending on the class of medical device. A PMS report is prepared for all products in class I (class I, Is, Im, Ir), while the PMS report for products in higher classes (IIa, IIb, and III) is called a periodic safety update report.
In general, the report on post-market surveillance must give an overview of gathered post-market surveillance data, analysis and evaluation of reported data, recommendations for any corrective and preventive actions to be taken, and conclusions on the benefit/risk determination. The level of detail included in this report depends on the risk class of the medical device and applicable regulatory requirements.
Considering MDR Article 85, for the PMS report for class I, it is necessary to prove that the PMS plan is realized as stated in Annex 3. For the periodic safety update report (PSUR), Article 86 requests that special attention should be paid to the conclusions of the benefit/risk determination, the main findings from post-market clinical follow up, the device sales volume, and the estimated size evaluation and other characteristics of the population using the device.
The defined content and time span for reviewing the PMS activities, taking into account the medical device class, as stated in the MDR, are presented in the following table:
Prepare a strong PMS system
When creating your PMS system, keep the following in mind: the device must show that it remains compliant when it is on the market. The biggest problem is setting up a strong PMS system for defining the data that are important for the medical device. The range of data to be monitored should be in direct proportion to the risk associated with the device based on its intended use. An effectively set-up PMS system indicates potential shortcomings in the design or production of your device and guides you as to how to address them.
But no matter how much work there is around post-market surveillance, at the same time, it allows you to prove your customer focus (end user), providing users with a consistently safe medical device for the right purpose.
To comply with all EU MDR requirements, use this helpful ISO 13485 & MDR Integrated Documentation Toolkit that provides all documents for medical device companies.



