

 Kristina Zvonar Brkic
Kristina Zvonar Brkic According to the definitions in the European Union’s Medical Device Regulation (MDR) (Article 2), “label” means any written, printed, or graphic information appearing either on the device itself, or on the packaging of each unit or on the packaging of multiple devices. The purpose of the labeling process is to identify a medical device and its manufacturer, and to communicate essential information on safety, use, and performance. It is intended for users of medical devices, both professionals and consumers, and for relevant third parties.
As we are all aware, the MDR brings many challenges for medical device manufacturers. One of them is labeling, with new requirements that ask for various kinds of information to be indicated on the labels of medical devices.
Labeling requirements under the new MDR
Compared to the MDD 93/42/EEC, there is a need for much more information on the labels under the EU MDR, because device safety and clinical effectiveness data is required to be shared transparently with users (both medical staff and patients/end users). All requirements regarding the information supplied with the medical devices are covered in Chapter III of Annex I, General safety and performance requirements, in the EU MDR.
There are two possible problems when trying to comply with the EU MDR labeling requirements. One is to be sure that all necessary symbols and information are covered. The other is the size of the label. As there will be many more symbols and data required, the big challenge will be how to fit it all on the label. During label design, keep the following in mind: the medium, format, content, legibility, and location of the label and instructions must match up with the technical knowledge, experience, education, or training of the intended user(s). Furthermore, instructions for use must be written in terms readily understood by the intended user and, where appropriate, supplemented with drawings and diagrams.
It is also good to know that you have a choice in format; labels can be provided in a human-readable format and may be supplemented by machine-readable information.
New elements on the label
Each medical device must have an indication that it is a medical device. If the device is intended for clinical investigation only, then the labels must contain the words “exclusively for clinical investigation.”
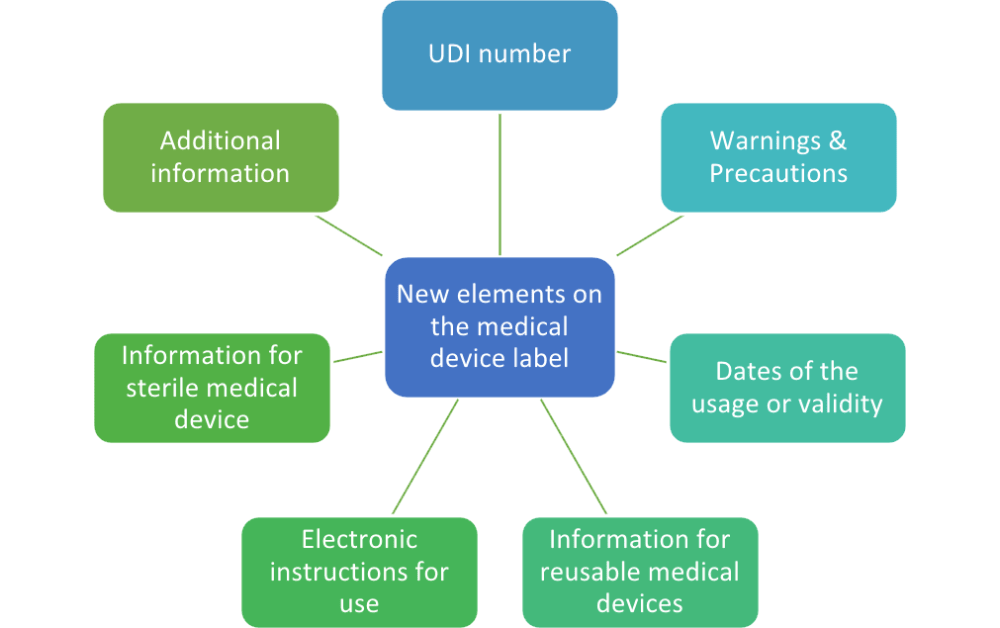
1) UDI number. So far, under the MDD, it was mandatory to include a lot number and/or a serial number on the label. The EU MDR introduces the term “UDI number,” which requires much more space on the label. Besides that, every active implantable device must have its own unique serial number, while other implantable devices will require a serial or lot number.
2) Warnings & Precautions. There is a request in Chapter III of Annex I, section 23.2, that all warnings relating to a device must be printed on the label. However, it is also stated that this information can be kept to a minimum, in which case more detailed information shall appear in the instructions for use, taking into account the intended users. The choice of which warnings need to be included is left to the manufacturer, but the best way is to use those warnings that request immediate attention.
3) Dates of the usage or validity. Where there is no indication of an expiration date, until which the medical device may be used safely, the date of manufacture must be present on the label. However, this date of manufacture may be included as part of the lot number or serial number, provided the date is clearly identifiable.
4) Reusable medical devices. Since the EU MDR has introduced a new class for reusable medical devices, there is a requirement to state the number of reprocessing cycles that are approved, along with any limitations.
5) Electronic instructions for use. It is possible to put a web address where electronic instructions for use can be found. This is especially convenient for those devices that are rather small, where there is physically not enough space to put all warnings and precautions on the label.
6) For sterile medical device. Besides all of the other required information (sterile method, symbol for sterile, date of validity), a description of the sterile barrier system is now required. The reasons for inclusion of such symbols are to mitigate specific risks with aseptic presentation, to comply with new legal requirements deriving from the EU MDR 2017/745, and to provide additional user benefits.
7) Additional information. The Medical Device Regulation requires all other specific information that further explains the product itself to be placed on the label. This includes information like:
- absorption rate
- blood or tissue derivate
- innovations like nanotechnology or computer software
- if there is a medicinal substance or tissue/cells
- presence of carcinogenic, mutagenic, or toxic for reproduction or endocrine-disrupting substance
New revision of ISO 15223
These increased labeling requirements are also reflected in the existing ISO standard for symbols. There is a new revision of the standard: ISO/DIS 15223-1:2020 Medical devices — Symbols to be used with medical device labels, labelling and information to be supplied — Part 1: General requirements – currently in the process of approval. There are completely new symbols that cover the new MDR requirements.
How to prepare
Achieving compliance with the MDR will naturally create labeling challenges for medical device manufacturers. Companies need to ensure that their current labeling system is fit for purpose. Therefore, the best way to prepare your MDR labels is to go through Annex I General safety and performance requirements, Chapter III, requirements 23.2 and 23.3, and to find out which of these requirements is applicable for your medical device. After you define that, you need to create the design of your label using symbols from ISO 15223-1:2020. Nevertheless, keep in mind that this design has to be flexible, because regulations are often changing, and you need to be able to respond to those changes very quickly.
To comply with all EU MDR requirements, use this helpful ISO 13485 & MDR Integrated Documentation Toolkit that provides all documents for medical device companies.



