

 Waqas Imam
Waqas Imam The design and development process for medical devices is not as simple as in other manufacturing or service industries. Design and development for medical devices has to deal with relevant regulations, product safety, and risk controls on product defects in addition to the usual application requirements, specification requirements, and end customer needs. The criticality of the design and development process is that if the medical device fails to meet regulatory design requirements, it won’t be able to reach the market.
An improper design is not merely a flawed design in a medical device product lifecycle; instead, it means the product can be dangerous and hazardous to patients and, subsequently, the product can be rejected by the regulatory body due to its (flawed) design, damaging the image of the medical device supplier. ISO 13485:2016 helps to carry out medical device design activities in effective and controlled manner.
Design and development process management
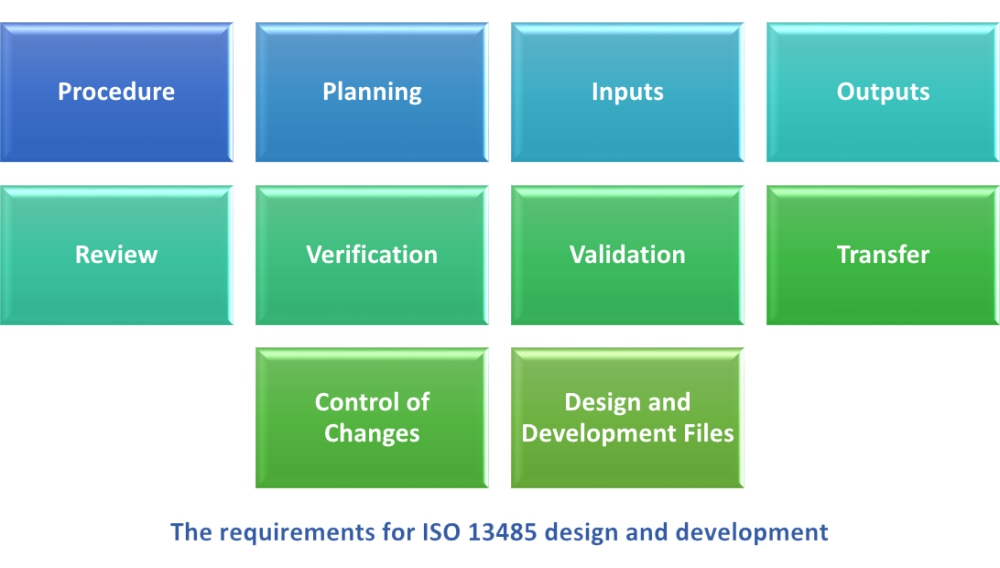
ISO 13485:2016 adds some new requirements within the scope of design and development, such as design and development transfer records and design files. The requirements for design and development are:
1) Procedure – The organization is required to document all the logical phases in design and development in a well-structured procedure, defining responsibilities for different activities, including approving authorities.
2) Planning – The planning phase is the most important phase of design and development, because proper planning can prevent unnecessary delays. In the planning phase the organization should identify the goal and objectives of the design and development of the product, the breakdown of major activities including risk management activities, the timeline of single activities and the whole project, and the allocation of resources needed in each phase of design and development (e.g., human resources needed in the review phase, i.e., review teams, devices that will be used for measurement and monitoring, etc.).
3) Inputs – As usual, it’s “garbage in – garbage out”; therefore, the quality of design and development inputs are crucial for producing the right outputs. What the organization should include as inputs are as follows:
- Intended application
- Usability requirements, for example, its application, preservation, handling, and maintenance
- Customer and end user requirements
- Physical features, attributes, and manufacturing feasibility
- Ergonomics and safety factors
- Risk control and risk mitigation techniques
- Past complaints, failure reports, and adverse events of similar products
- Relevant regulatory, legal, and statutory needs and appropriate standards
- Sterilization requirements and servicing needs
- Economic study and costing feasibility
4) Outputs – The organization can produce design outputs in the following forms:
- Raw materials, component parts, sub-assemblies, and finished device specifications in drawings
- Manufacturing process and environmental specifications
- Procedure for quality assurance that explains acceptance criteria
- Product identification, traceability, manufacturing, packaging, and inspection procedures
- Documentation for submission to the regulatory authorities where devices will be marketed
- Design history file to demonstrate design was verified and validated
5) Review – The design review is a detailed step that addresses a number of manufacturing and customer concerns. For example, the organization needs to demonstrate whether the design meets product requirements or not, whether the device design exhibits compatibility with processing capabilities or not, whether safety concerns are addressed or not, whether it is environmentally friendly or not, and whether materials, facilities, components, and service elements are adequate or not. Design reviews are normally done in a meeting, and minutes should be maintained.
6) Verification – Design verification is a mandatory requirement. It ensures that design outputs meet the specified requirements of inputs. The organization can verify designs with the help of tests (lab tests, chemical analysis, etc.), substitute calculations, comparing proven designs, inspections, and reviews of documents like specifications records, drawings, procedures, plans, reports, etc.
7) Validation – Design validation is a step that comes after design verification. It is a phase that makes sure that the medical device conforms to end user requirements and the application. Validation is done on samples from initially produced lots. The product is validated in simulated conditions where its actual performance is tested (e.g., clinical testing of medical devices). Records of design validation must be maintained.
8) Transfer – An organization must document a procedure to transfer design and development outputs to manufacturing. This is not just handing off and taking over of design from product development to the manufacturing department. Rather, it means that product development has made sure that the design can be translated to production and records of such transfer are maintained.
9) Control of Changes – The procedure for design and development of the medical devices should include a mechanism to control design and development changes. A design change can be needed at any time based on review, verification, validation, complaints, risk mitigation, manufacturing issues, etc. Prior to change enforcement, it should be reviewed, verified, validated, and approved against design inputs and requirements.
10) Design and Development Files – The organization should maintain a design and development file for each medical device design. The file may include reference records of conformity to design requirements, records of review, verification, validation, and changes.
Helping organizations with an effective design and development process
ISO 13485:2016, with its new requirements for design and development, has made the process safer for implementing organizations. In the review and validation phase, the design manufacturing process is critically reviewed at every step for shortcomings. Design flaws are thus corrected on factors like device safety, market competitiveness, regulatory consent, user satisfaction, functional usability, and profit returns, and eventually, improvements are incorporated into the approved design.
ISO 13485 design and development guidelines play an important role in the medical device lifecycle; they add value for the end users, patients, and hospitals, and fulfill the needs of regulators. What else could medical device manufacturers and suppliers look for?
To implement ISO 13485 easily and efficiently, use our ISO 13485 Documentation Toolkit that provides step-by-step guidance and all documents for full ISO 13485 compliance.



