

 Kristina Zvonar Brkic
Kristina Zvonar Brkic ISO 13485:2016 is an international quality management standard for manufacturers of medical devices. It specifies the requirements for a quality system in which the manufacturer needs to demonstrate its ability to deliver safe medical devices. The primary goal of ISO 13485 is to establish a system that is fully capable of meeting applicable legal and quality system requirements.
Benefits of ISO 13485 for manufacturing companies
- Possibility of contracting with larger companies;
- Ensuring reliability and quality of the medical device in different countries;
- Proof of compliance with legal, regulatory, contractual, and marketing requirements;
- Demonstrating product safety to the patient/end user.
ISO 13485 is a stand-alone standard. It relies heavily on the ISO 9001:2008 structure, but it includes certain specific requirements for medical devices, such as risk analysis, sterile production, rework, and traceability. An “ISO 13485-certified company“ means an organization that has implemented an ISO 13485 Quality Management System (QMS) and has successfully met all the applicable requirements (considering the nature of the medical device), audited by the certification body. Read this article to learn how obtaining ISO 13485 certification can benefit manufacturing companies.
How is ISO 13485 applicable for manufacturing companies?
ISO 13485 is intended for any organization partially or fully involved in the medical device lifecycle, regardless of the organization’s size or type, or the type of medical device. The main purpose of an ISO 13485 QMS is to prove the manufacturer’s commitment to the safety, effectiveness, and performance of medical devices.
Which manufacturers are involved in the medical device lifecycle? The ones that are involved in the following processes related to medical devices: design and development, production, storage and distribution, installation, and servicing, as well as the provision of associated activities (e.g., technical support).
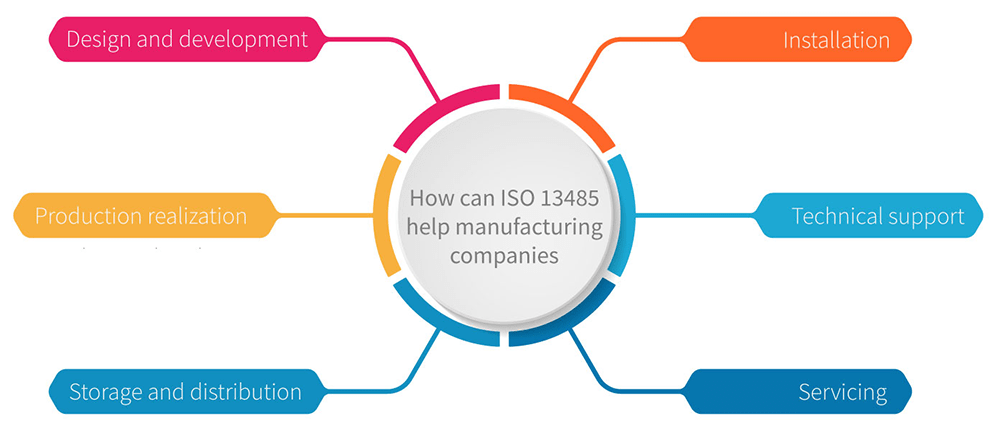
Design and development process. The point is that medical devices must be designed in a proper manner to ensure the safety of patients and users, and they must perform in accordance with their intended use. Introducing ISO 13485 into your design and development process will guide you to define exactly and in detail what your input parameters are, how you will collect them, and who is responsible for each input parameter. It will also give you control over design changes, so you know at all times at what stage your new medical product development is.
Furthermore, ISO 13485:2016 requires you to ensure traceability throughout the design and development process. This is important because anyone can quickly gain insight into what stage the design is, check out some specific designs or risky elements, or be up to date with certain changes. Properly implemented traceability allows you agility in managing the design and development process.
Also, the ISO 13485:2016 design and development requirements instruct you to monitor product risks at the very beginning of the design, which significantly increases the safety of the final product.
ISO 13485 requires the company to constantly review, verify, and validate each phase of a development project. This ensures that there is complete control over the development process and ensures that the final product is safe.
Production realization is the main point in ISO 13485:2016. The goal of every medical device manufacturer is for the production process to be under maximum control so that the product that comes out is in accordance with customer requirements, is safe to use, and is fit for the expected intended use. ISO 13485 allows easy definition of production processes, control steps, traceability, process performance monitoring, and performance monitoring of the finished product.
The following elements are also necessary to maintain proper production: defining requirements for infrastructure and equipment, maintaining and monitoring infrastructure and equipment, defining ambient conditions for production, defining the required cleanliness, etc. ISO 13485 allows you total control over the production process, which is essential for the production of a safe medical device.
Storage and distribution. It is very important to properly store and protect material, parts, and medical devices during the complete lifecycle: production, storage, handling, and distribution. ISO 13485:2016 asks you to protect product from any alteration, contamination, or damage. This may include monitoring temperature, humidity, and any contamination. As in the warehouse, you must also protect your product during its transport to the customer. This means that if your product requires refrigeration, you must ensure that the product travels at the correct temperature during transport. Requirements and criteria must be prescribed, and there must be evidence of their monitoring.
Installation. If a medical device requires installation at the point of use (at the customer site), then installation requirements and specifications must be defined. There should be established installation protocol, acceptance criteria, and an installation report/record. It is very important that the manufacturer of the medical device delivers a fully valid medical device ready for use.
Servicing. Certain medical devices require servicing, for which requirements and specifications must also be defined. It is very important to ensure that, once serviced, the product still meets the basic requirements of the medical device. The performance and safety of the product must not be changed after the service. Of course, there must be some service records and those records must be maintained.
Technical support. If the production of a medical device requires other technical support, the company providing that support is expected to comply with ISO 13485. Technical support can be, for example, sterilization service, or the production service of a particular part that needs to be incorporated into your medical device. The benefit of introducing ISO 13485 in such companies is that the medical device manufacturer has complete traceability of the entire manufacturing process. Ultimately, the manufacturer of a medical device is solely responsible for that medical device once it enters the market.
Should you go for ISO 13485 or some other standard?
A common question is: Do manufacturers need to go for ISO 13485, and do they need a Quality Management System at all or is it their own choice? Considering the EU market, the following Medical device regulation MDR 2017/745 Article 10 – “General obligations of manufacturers” states that manufacturers of devices, other than investigational devices, must establish, document, implement, maintain, keep up to date, and continually improve a Quality Management System that shall ensure compliance with this Regulation.
In MDR Article 8 – “Use of harmonized standards”, is stated that to be in compliance with the MDR, all medical devices must be designed and produced in accordance with standards published in the Official Journal of the European Union, so-called “harmonized standards.” Currently, the only standard that covers Quality Management Systems on the list of harmonized standards is ISO 13485:2016. Therefore, compliance with ISO 13485:2016 provides presumption of conformity with the basic QMS requirements for CE marking under the MDR. To learn more, read this article: How can ISO 13485 help with MDR compliance?
The importance of ISO 13485 was also recognized by the U.S. Food & Drug Administration (FDA). In December 2018, this organization issued a proposed rule to harmonize U.S. Quality System Regulations (21 CFR 820) with ISO 13485 and make ISO 13485 mandatory.
To learn about the relationship of regulatory authorities with ISO 13485 in other markets, see the following article: List of worldwide regulations that require implementation of ISO 13485.
ISO 13485 benefits for manufacturing companies
Benefits for the companies that implement ISO 13485:2016 are as follows:
- Possibility of contracting with larger companies – many large medical device companies prefer to work with suppliers who are ISO 13485 certified. Large companies are responsible for ensuring that their subcontractors comply with ISO 13485, which means that subcontractors who are already certified are likely to have priority.
- The ISO 13485 standard was created to ensure that medical devices in different countries show the same reliability and quality. If you are considering exporting a product, ISO 13485 certification can give you an advantage.
- By implementing ISO 13485, the organization is proving that it meets all applicable legal, regulatory, contractual, and marketing requirements.
- Demonstrating product safety to the patient/end user – ISO 13485 instructs manufacturers to produce a safe product using a risk-based approach. The organization must analyze the entire production process from the point of view of risk in order to reduce risk to a minimum.
Ensuring control over the manufacturing process
With the introduction of ISO 13485: 2016, manufacturers will organize their processes so that they know at all times who did what, with what, where the results are, and who is responsible for performing each stage of production. It will give them control over the entire product process, but also control over checking the quality of the medical products. ISO 13485 is a tool that guarantees them that the medical device is manufactured in a safe way for both the patient and the user, and that is what is most important.
To implement ISO 13485 easily and efficiently, use our ISO 13485 Documentation Toolkit that provides step-by-step guidance and all documents for full ISO 13485 compliance.




